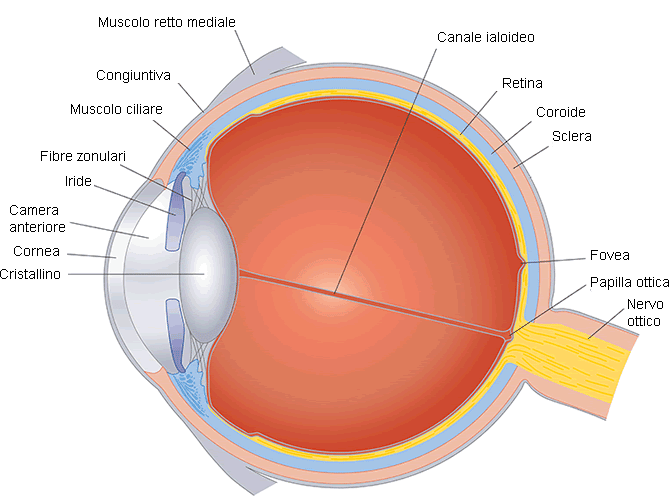
La retina è la membrana più interna del bulbo oculare, un tessuto di origine nervosa fondamentale per la suddivisione e la trasduzione delle immagini refratte dalla struttura ottica del nostro occhio. Riveste quasi tutta la parete interna dell’occhio, coprendo circa 270° della sua sezione. Vediamo quali sono le distrofie retiniche più diffuse.
La retina umana ha uno spessore variabile da circa 0,4 mm nella parte centrale a 0,1 mm nella parte più marginale ed è formata da cellule fotorecettori sensibili alle onde luminose. I coni permettono la visione a colori, ma sono attivi unicamente quando la luce è intensa, ovvero sono coinvolti nella visione diurna. I bastoncelli sono attivi nella visione monocromatica in condizioni di luce soffusa o crepuscolare.
Questo complesso apparato nervoso funziona come un fototrasduttore (trasforma l’energia luminosa in potenziale elettrico): capta gli stimoli luminosi e li converte in stimoli nervosi, segnali bioelettrici, che poi vengono inviati al cervello attraverso le fibre del nervo ottico. Molti studiosi ritengono che la retina sia parte periferica del cervello e non un organo esterno.
Le distrofie retiniche rappresentano un gruppo di malattie rare, a prevalenza di origine genetica, la cui reale genesi ad oggi non è nota. Colpiscono la retina, la coroide ed a volte entrambe, conducendo a delle alterazioni progressive sia anatomiche che funzionali. Alterazioni morfo funzionali che conseguono a delle modificazioni introdotte nel trofismo retinico normale. Data l’importanza e la complessità dell’apparato retinico, è fondamentale approfondire lo studio della sua degenerazione e del suo amorfismo.
Ad oggi vengono clinicamente riconosciute otto forme diverse (con alcune varianti) di distrofia retinica con una incidenza del 3% circa della popolazione. Anche i modelli di ereditarietà sono diversi come lo sono i sintomi, l’età di insorgenza ed il livello di gravità dell’anomalia e del suo effetto sulla funzionalità visiva dell’individuo. Le otto forme di distrofie retiniche identificate sono:
A scopo diagnostico ed in vista di un eventuale approccio terapeutico, queste distrofie vengono suddivise in due macro categorie: le distrofie retiniche di origine maculare e le distrofie retiniche di origine periferica. Quelle di origine maculare originano nella zona centrale della retina, detta anche macula che sovrintende l’acuità visiva. Le distrofie di questa natura alterano la misura della visione in una età più precoce rispetto a quelle di origine retinica periferica, le più comuni sono la Malattia di Stargardt e la Malattia di Best. Le distrofie retiniche di origine periferica determinano una riduzione progressiva del campo visivo periferico fino a compromettere la porzione centrale della visione; le più comuni sono la retinite pigmentosa e la coroideremia.
In ordine di sintomatologia, i pazienti colpiti da una forma di distrofia retinica purtroppo sperimentano un ampio ventaglio di esperienze. Fondamentalmente legati al tipo di morfologia della retina, questi sintomi influenzano conseguentemente la funzionalità del tessuto nervoso. In generale si possono classificare questi segnali degenerativi come alterazioni del campo visivo, cecità od ipovisione, cecità notturna, metamorfopsia (percezione distorta o deformata degli oggetti, che rivela alterazioni nell’area centrale della retina) e percezione alterata dei colori.
La cecità retinica congenita, compare nei primi sei mesi di vita, ed in alcuni casi può essere progressiva. Comporta una cecità od ipovisione, associate a nistagmo. Nella maggior parte dei casi lo sviluppo psicomotorio è nella norma e non vi sono malformazioni o disfunzioni associate a carico di altri organi e apparati.
La distrofia dei coni-bastoncelli rappresenta una patologia prettamente ereditaria; comunque si osservano anche numerosi casi a prima vista isolati. Questa distrofia viene contraddistinta dalla degenerazione dei coni e dei bastoncelli e dalla consequenziale alterazione della capacità visiva. Un approfondimento motivato dalla natura ereditaria di questa malattia, sulla traslocazione cromosomica, ha portato nel 1986 a trovare un caso di distrofia dei coni legato a ritardo mentale e dimorfismi facciali.
La distrofia ialina della retina è contraddistinta dalla perdita graduale della vista o cecità notturna e segni oculari quali la liquefazione del corpo vitreo, la retinoschisi maculare e l’atrofia e la pigmentazione periferica dell’epitelio pigmentato della retina. L’elettroretinogramma risulta estinto o marcatamente anomalo.
La malattia di Best colpisce la macula, parte centrale della retina responsabile della visione fine e dettagliata e della percezione dei colori. Rappresenta una forma ereditaria di degenerazione maculare che è caratterizzata dalla perdita della visione centrale.
La cisti retinica congenita (distrofia vitroretinica) figura una malattia ereditaria che viene diagnosticata durante l’infanzia. Causa la perdita progressiva della visione centrale e periferica. È una malattia rara prevalentemente sconosciuta che colpisce quasi esclusivamente il sesso maschile. Altri sintomi ad insorgenza precoce sono lo strabismo ed il nistagmo o la presenza di movimenti oculari anomali. Il deficit visivo è causato da una fenditura della retina che si divide in due strati: gli spazi creati dalla fenditura retinica vengono spesso occupati da vescicole e da vasi sanguigni, che rompendosi generano sanguinamento nel corpo vitreo.
La malattia di Stargardt è la più comune forma di degenerazione maculare giovanile: è caratterizzata dalla riduzione della visione centrale e dalla conservazione della visione periferica; è una malattia rara e con una incidenza sconosciuta, diagnosticata in genere dai 20 anni in giù.
La retinite pigmentosa rappresenta un insieme di malattie retiniche la cui caratteristica è una progressiva degenerazione delle cellule fotorecettoriali ed un conseguente progressivo deficit visivo. La sintomatologia clinica varia secondo il tipo di cellule coinvolte e include la cecità notturna, la perdita della visione periferica e la perdita della capacità di distinguere i colori.
La retinopatia punteggiata albescente delinea una forma atipica e progressiva di retinite pigmentosa con modalità di trasmissione autosomica recessiva della quale il gene responsabile non è noto. I sintomi caratteristici sono l’emeralopia e il difetto campimetrico, con una progressiva visione tubolare. È caratterizzata dalla presenza regolare, su tutto il territorio retinico, di chiazzette biancastre che precedono o coesistono con la tipica pigmentazione della retinite pigmentosa.
Come accennato in precedenza, le cause delle anomalie che colpiscono la retina sono per la maggior parte da ricercarsi nelle anomalie genetiche che possono trasmettersi su base ereditaria. Nonostante le attuali conoscenze nel campo della genetica, resta tuttavia molto difficile isolare la causa diretta di gran parte di queste malattie. Sono delle anomalie piuttosto rare e come tali, rendono lo studio molto difficile per la complessità dei campioni statistici e per le scarse ripetizioni genetiche da esaminare.
Tuttavia, nonostante ci si muova in un campo piuttosto sconosciuto, si è riusciti ad isolare dei geni specifici o delle sequenze precise responsabili dirette di alcune alterazioni. In questo modo è stato possibile creare protocolli terapeutici adatti, e persino mettere a punto cure specifiche da somministrare a pazienti recettivi riscontrando anche dei successi. Questa strada è percorribile al punto che l’AIFA ha autorizzato trattamenti specifici per specifiche patologie retiniche e grazie a questi percorsi terapeutici è stato possibile ridare speranza e la vista a pazienti in giovane età.
Nonostante la rarità e le peculiarità di queste malattie, è possibile diagnosticare le distrofie retiniche con differenti tecniche. Spesso l’esame diretto ed accurato del fondo oculare può fornire un riferimento diagnostico molto affidabile.
In altri casi, la diagnostica per immagini complementare può essere significativamente utile per comprendere lo stato della malattia: l’autofluorescenza o la tomografia a coerenza ottica, permette un esame approfondito della morfologia oculare; elettroretinogramma, elettrooculogramma o potenziali evocati visivi, sono test che permettono invece di approfondire gli aspetti funzionali. Con le conoscenze odierne, in caso di dubbio diagnostico oppure per fornire una prova di conferma definitiva, si può eseguire un test genetico e mediante questo rilevare la presenza del gene o dei geni mutati responsabili della specifica malattia.
Nel caso di una cecità retinica congenita si integrano esami preliminari ed esami più approfonditi: un esame del fondo oculare ed una valutazione neuroftalmologica completa viene affiancata a PEV, ERG; solitamente si effettua anche uno studio RMN dell’encefalo per escludere malformazioni del sistema nervoso centrale. A questi esami, si aggiunge lo studio delle mutazioni genetiche individuate a scopo di ricerca.
Per la distrofia dei coni-bastoncelli il sospetto clinico si può confermare attraverso l’esame del fundus oculi, l’elettroretinogramma, la fluorangiografia retinica e l’esame campimetrico. La diagnosi della distrofia ialina della retina resta clinica e strumentale: attualmente non esiste un test genetico di conferma diagnostica. Sebbene l’età di esordio per la malattia di Best sia variabile, generalmente viene diagnosticata durante l’infanzia o l’adolescenza. Negli stadi iniziali della malattia, una cisti color giallo lucente si forma sotto l’epitelio pigmentato della retina, al di sotto della macula; l’esame funduscopico rivela che la cisti ha una forma ovalare. Dal punto di vista della diagnosi genetica, è stato mappato un gene sul braccio corto del cromosoma 11 (locus 11q13) associato a questa malattia.
La cisti retinica congenita prevede una diagnosi clinica e fundoscopica, in questo momento non sono disponibili test molecolari di conferma diagnostica. Anche per la malattia di Startgardt la diagnosi praticabile è clinica e fundoscopica. La retinite pigmentosa può essere diagnosticata con il metodo clinico strumentale, è possibile anche una analisi molecolare per la ricerca di mutazioni note. Infine, anche per la retinopatia punteggiata albescente la diagnosi è di tipo clinico e strumentale; un segno tipico è che l’ERG risulta alterato o estinto. Al momento non esiste un test genetico per la conferma diagnostica.
Come ampiamente osservato, le distrofie retiniche rientrano per lo più nel campo delle malattie rare, quindi i trattamenti terapeutici sono abbastanza limitati. In generale, attualmente non c’è nessun trattamento disponibile per le distrofie della retina valido per tutti i casi. Gli studi ed i trials clinici rammentano che sia la terapia genica che quella cellulare potrebbero avere un ruolo terapeutico, ma molto resta legato ai singoli casi ed alle disponibilità del momento. A titolo di esempio, in alcuni casi avanzati di retinite pigmentosa, esiste la possibilità di un impianto sopra la macula (ARGUS II) che migliora l’acuità visiva del paziente dalla percezione della luce alla localizzazione degli oggetti.
Per la cecità retinica congenita attualmente non è disponibile una terapia unica e risolutiva, tuttavia la ricerca si sta muovendo in più direzioni: dal trapianto di cellule retiniche, alla terapia farmacologica (si punta a migliorare il residuo visivo nei pazienti con funzionalità dei bastoncelli residua); si sta sperimentando il trapianto di cellule staminali, o l’impianto di microchips. Degno di nota, un recente studio in modelli murini con mutazione REP65 ha dimostrato miglioramenti a breve termine a livello neurofisiologico con implementazione dietetica di Vitamina A.
Anche per la distrofia dei coni-bastoncelli non esiste alcuna terapia unica risolutiva del quadro clinico. Nel caso in cui l’acuità visiva del paziente è ridotta, possono essere utili gli ausili per ipovedenti; si può ricorrere a lenti protettive per la luce. Data la natura della malattia, è utile ricorrere ad una consulenza genetica sia per genitori che per il paziente o i suoi figli. Va notato che, sebbene siano stati fatti numerosi passi in avanti in campo diagnostico e terapeutico, oggi non è disponibile alcun trattamento causale per le varie forme di distrofie retiniche ereditarie.
Purtroppo, per la distrofia Ialina della Retina ad oggi non esiste alcun trattamento causale della malattia. Si può ricorrere ad un trattamento profilattico delle rotture retiniche asintomatiche per prevenire il distacco di retina. Anche nel caso della malattia di Best le conoscenze attuali non permettono un trattamento causale della malattia. I pazienti affetti da questa retinopatia possono ricorrere ad ausili per l’ipovisione e ad esercizi di training per l’orientamento spaziale. Al momento non è disponibile un trattamento causale nemmeno per la cisti retinica congenita. Si può trattare con un opportuno intervento chirurgico il distacco retinico e si possono implementare ausili per l’ipovisione e training per l’orientamento spaziale.
Benché ad oggi non sia disponibile un trattamento causale per la malattia di Startgardt, anche in questo caso, il paziente può beneficiare di ausili per l’ipovisione e di training per l’orientamento spaziale. Anche la retinopatia punteggiata albescente ad oggi non prevede protocolli terapeutici causali. Valersi di specifici farmaci come vasodilatatori, biostimolanti, aminoacidi essenziali, non ha prodotto riscontri positivi apprezzabili. L’efficacia resta transitoria, probabilmente grazie all’aumento transitorio del metabolismo locale
Nel caso del trattamento della retinite pigmentosa, sono stati predisposti molti protocolli terapeutici: si punta soprattutto ad avere un effetto sul metabolismo del fotorecettore mediante la vitamina A o gli antocianosidi, tuttavia nessun trattamento terapeutico si è dimostrato in grado di guarire o di modificare la progressione della malattia. Comunque, la terapia con ossigeno potrebbe essere di notevole utilità poiché l’ossigeno influisce positivamente riducendo il tasso di morte cellulare dei fotorecettori.
Sono stati approfonditi numerosi modelli animali di retinite pigmentosa che hanno migliorato la comprensione dei meccanismi patogenetici della malattia e dei possibili presidi terapeutici. Nel particolare, la terapia genica potrebbe rivelare una sensibile svolta: fattori specifici di crescita e supplementazioni vitaminiche potrebbero rallentare o addirittura interrompere i processi di degenerazione dei fotorecettori. Ulteriormente, l’impiego di cellule staminali potrebbe sostituire le cellule perdute con un possibile recupero funzionale.
Da quanto considerato finora, si comprende chiaramente quanto le distrofie retiniche rappresentino un problema serio per la Medicina moderna e che richiedano un approccio terapeutico molto scrupoloso. Se poi si considera che gran parte di questi disturbi affligge pazienti, molti dei quali bambini o nella fascia di età under 20, particolarmente sensibili agli effetti quotidiani della patologia, ci si rende conto da subito che è necessario profondere più delle solite energie nella ricerca medico-scientifica.
Come menzionato, diverse di queste patologie retiniche non prevedono una cura causale e non possono essere prevenute o curate dovutamente per limitare i danni alla vita del paziente. Spesso si può provvedere a limitare questi danni, fornendo terapie di supporto o strumenti che contengano i disagi conseguenti alla perdita o alla menomazione della vista.
Tuttavia, in questo scenario piuttosto scuro, si inserisce un recente successo raggiunto da diverse equipe mediche che sono state in grado di restituire la vista ad alcuni bambini mediante trattamenti mirati e terapie personalizzate. Nello specifico, alla Clinica Oculistica dell’Università degli Studi della Campania “Luigi Vanvitelli”, dieci bambini affetti da distrofia retinica ereditaria sono stati trattati con terapia genica ed il 25 ottobre 2021 sono stati resi noti i risultati estremamente incoraggianti e positivi di questo trattamento.
In buona sostanza è stato un eccellente preludio a quanto avvenuto più di recente. Il 19 settembre del 2022 un Comunicato Stampa riportava che la collaborazione tra il Policlinico Gemelli e l’Ospedale Pediatrico Bambino Gesù ha reso possibile ridare la vista a due fratellini, affetti da distrofia retinica ereditaria, grazie alla terapia genica. La notizia è stata accolta con chiaro entusiasmo dalla comunità scientifica e dai media in generale, il comunicato in parte recita:
Un fratellino e una sorellina di 8 e 3 anni, affetti dalla stessa forma di distrofia retinica ereditaria, hanno riacquistato importanti capacità visive in seguito al trattamento con terapia genica.
Gli interventi sono stati eseguiti in collaborazione dalle unità di Oculistica della Fondazione Policlinico Universitario Agostino Gemelli IRCCS e dell’Ospedale Pediatrico Bambino Gesù, nell’ambito di un progetto avviato nel 2021 per la gestione comune di pazienti pediatrici e adulti affetti da degenerazioni retiniche ereditarie. La bambina che ha riacquistato la vista è la più giovane paziente in Italia ad aver ricevuto questo trattamento”.
La terapia genica è stata approvata nel 2021 dall’AIFA ed agisce sul gene RPE65 responsabile della produzione di una proteina chiave nel processo di conversione della luce in segnale elettrico nella retina. I fratellini trattati erano affetti da Amaurosi congenita di Leber e le mutazioni sulle due copie del gene sono tanto rare da interessare circa 1 persona su 200 mila nel mondo. In buona sostanza, la terapia consiste in una singola iniezione nello spazio sottoretinico di entrambi gli occhi di una copia funzionante del gene RPE65. Il gene sano è veicolato all’interno delle cellule da un adenovirus associato, con patrimonio genetico modificato, che agisce come vettore. Una volta nelle cellule, la copia funzionante del gene è in grado di ripristinare la capacità visiva del paziente in modo significativo e duraturo.
Il percorso di follow-up post intervento, completato a febbraio del 2022, ha evidenziato un significativo miglioramento di tutti i parametri visivi soggettivi, cioè l’acuità visiva (capacità di discriminare un dettaglio spaziale), il campo visivo (capacità di vedere perifericamente) e la visione crepuscolare. Lo stesso vale per i parametri oggettivi attraverso test specifici quali l’FST (test che valuta la sensibilità dei coni e bastoncelli, la popolazione cellulare retinica che ci consente di vedere).
Questo risultato entusiasmante rappresenta una risposta terapeutica concreta per la prevenzione e la cura di una parte delle distrofie retiniche, apre uno scenario nuovo sulle future strategie di approccio e di cura alle degenerazioni della retina e ci concede di sperare in un futuro nel quale la tecnologia genetica, il supporto di microchips sottocutanei, il sostegno dei percorsi di training per la percezione e l’orientamento spaziale e la disponibilità di supporti per ipovedenti sempre più avanzati, rendano la vita quotidiana di chi è affetto da distrofia retinica, sempre più agevole e vicina alla normalità.
Concludiamo con le parole lungimiranti ed incoraggianti del Prof. Stanislao Rizzo, professore ordinario di Oculistica presso l’Università Cattolica e direttore della UOC di Oculistica del Policlinico Universitario A. Gemelli IRCCS:
Operare pazienti in giovanissima età e ridare loro la vista è qualcosa che travalica la nostra vita professionale.
A cura di Francesca Maria Iervolino
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}