Formazione di aggregati proteici nelle patologie neurodegenerative

Le patologie neurodegenerative sono un insieme eterogeneo di malattie che interessano il sistema nervoso centrale, causando una morte cellulare cronica e selettiva dei neuroni. A seconda del tipo di deterioramento neuronale in atto, si possono avere sintomi molto diversi, anche se in generale possono comprendere deficit cognitivi, demenza, alterazioni motorie, disturbi comportamentali e psicologici. Ovviamente molto dipende dall’area del cervello interessata e dal tipo di neurone che viene colpito. La progressione causata dalla morte dei neuroni è un processo irreversibile, anche per il fatto che la neurogenesi nell’adulto è molto limitata, ma è comunque presente.

Attualmente non esistono trattamenti farmacologici in grado di arrestare del tutto questi processi, tuttavia in molti casi si riesce a rallentarli. Tendenzialmente gli stadi iniziali della malattia sono asintomatici e quindi difficilmente diagnosticabili. Questo dipende dal fatto che i primi sintomi compaiono solo in seguito a un danno neuronale consistente. Per molte di queste patologie le cause scatenanti non sono ancora del tutto chiare anche se si ritiene sia importante il coinvolgimento di più fattori che concorrono per dare inizio alla neurodegenerazione tra cui quelli genetici e ambientali.
La peculiarità di alcune patologie neurodegenerative è la comparsa di corpi formati solitamente da agglomerati proteici che vanno ad intaccare le funzioni cerebrali, partecipando così, insieme alla morte neuronale, allo sviluppo della malattia e alla comparsa dei sintomi. Queste formazioni sono diverse a seconda del caso e si possono ritrovare sia a livello cellulare che extracellulare.
L’amiloidosi
L’amiloidosi è una patologia causata dall’accumulo di materiale proteico con ridotto peso molecolare ed insolubile in sede extracellulare. La sostanza è definita per l’appunto amiloide per la sua proprietà simile a quella dell’amido di reagire con lo iodio. Mentre inizialmente si considerava una sostanza del tutto amorfa, ora sappiamo che le proteine si organizzano in strutture a β-foglietto, motivo per cui l’amiloidosi è spesso rinominata come β-fibrillosi. Le fibrille in questione hanno una composizione chimica molto varia poiché a seconda della patologia in questione cambia la proteine precursore che si deposita.
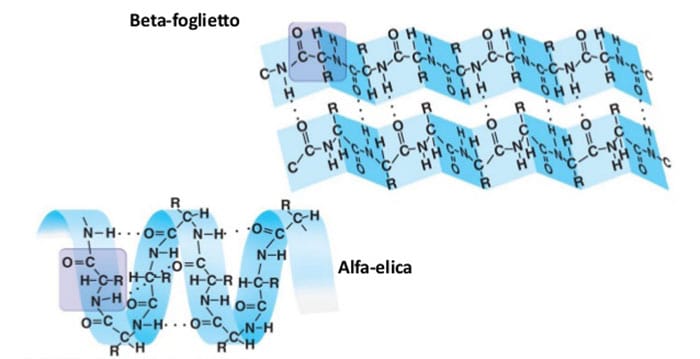
Questo a causa di una mutazione nella proteina oppure nell’errore di qualche enzima proteolitico che la dovrebbe trasformare. Oltre alle proteine disposte a β-foglietto si possono ritrovare esternamente carboidrati complessi e la proteina SAP (serum amyloid P component), mentre più verso la parte interna si trova il nucleo di componente AP (amyloid P component), una glicoproteina globulare, insieme ad aggregati di condroitinsolfato ed eparansolfato. L’importante è la struttura a β-foglietto che permette a proteine di solito solubili di aggregarsi in composti insolubili ed acquisire un’elevata resistenza ai processi proteolitici, diventando difficilmente degradabili dall’organismo.
La sostanza amiloide si può depositare a livello di organi diversi, accumulandosi e causando danni specifici a seconda della patologia. Si conoscono attualmente circa 41 malattie differenti associate ad amiloide. A livello cerebrale questa sostanza è la principale componente delle placche senili, o druse, spesso presenti nell’anziano, in particolare nei casi di demenza.
Aggregati proteici nelle patologie neurodegenerative: le placche senili
Le placche senili sono formazioni extracellulari composti da sostanza amiloide circondata da detriti neuronali, perlopiù frammenti di assoni. Sono riscontrabili in diverse regioni del sistema nervoso centrale tra cui l’encefalo, l’ippocampo, il giro del cingolo e nelle cortecce associative delle regioni frontali e temporo-parietali. Le placche senili sono le principali caratteristiche riscontrabili a livello microscopico in alcune patologie neurodegenerative come la demenza senile, il morbo di Alzheimer e l’angiopatia amiloide cerebrale.
I grovigli neurofibrillari
Le placche senili presenti nei pazienti affetti da Alzheimer o angiopatia sono accompagnate anche dagli ammassi neurofibrillari. Si tratta di formazioni intracellulari composte da filamenti elicoidali presenti nel citoplasma che avvolgono il nucleo dei neuroni, anche queste formazioni sono insolubili e difficilmente soggette alla proteolisi. La comparsa di questi grovigli è dovuta ad un iperfosforilazione della proteina Tau, che incrementa il montaggio dei microtubuli degli assoni, modificandone quindi il citoscheletro.

Aggregati proteici nelle patologie neurodegenerative: la proteina prionica
Tutte le encefalopatie spongiformi trasmissibili sono patologie neurodegenerative associate alla proteina prionica che causa demenza e disturbi neurodegenerativi. Tale proteina è normalmente presente in molti tipi di cellule del sistema nervoso centrale, del sistema linfatico e dell’apparato digerente. Le forme mutate di questa proteina assumono una disposizione a β-foglietto che, esattamente come nel caso dell’amiloide, consente loro di aggregarsi, di diventare insolubili e di resistere ai processi proteolitici. Infatti il risultato è il medesimo, ossia la formazione di strutture simili alle placche senili presenti nell’Alzheimer, anche se spesso sono accompagnate da queste ultime.
Tra le patologie da prioni si possono menzionare l’insonnia familiare fatale, la sindrome di Gerstmann-Sträussler-Scheinker, la malattia di Creutzfeldt-Jakob e la sua variante che tutti conoscono come il morbo della mucca pazza. Le patologie prioniche possono avere un origine genetica oppure un origine infettiva, sono infatti le uniche proteine ad avere una capacità infettante. Ogni proteina prionica infettiva è in grado di far cambiare conformazione alle stesse proteine normalmente presenti nell’organismo, facendo loro assumere la tipica struttura a β-foglietto con quello che viene definito un meccanismo a stampo.

I corpi di Lewy
I corpi di Lewy sono aggregati proteici anormali che si formano nelle cellule nervose, appaiono come delle masse di forma sferica che spostano altri componenti cellulari. Si tratta di inclusioni citoplasmatiche eosinofile composte da aggregati di α-sinucleina e di ubiquitina che formano un nucleo denso circondato da fibrille. Si differenziano in due varianti morfologiche, quelli sottocorticali presenti nel tronco encefalico e quelli corticali che appaiono invece meno definiti.
La presenza di queste formazioni è associata a vari tipi di patologie neurodegenerative, tra cui le principali sono il morbo di Parkinson e la demenza da corpi di Lewy. Occasionalmente possono essere presenti in pazienti con taupatie, come la malattia di Pick o la degenerazione cortico-basale, inoltre sono associati ad una variante del morbo di Alzheimer, la sindrome di Hallervorden-Spatz. Esistono anche formazioni simili ai corpi di Lewy presenti nelle cellule gliali anziché nei neuroni, chiamate inclusioni citoplasmatiche gliali, riscontrabili in persone affette da atrofia multi-sistemica.