genoma
Un nuovo studio ha rivelato che la recente mappatura del genoma porta ad informazioni più dettagliate in merito alla ricerca sulle malattie; infatti, il sequenziamento dell’intero genoma offre uno sguardo senza precedenti alla composizione e al comportamento del cancro, ad esempio nel linfoma di Hodgkin. Questa recente scoperta potrebbe portare ad una migliore comprensione dello sviluppo e del trattamento di questo e di altri tumori.
Il genoma, in particolare il cariotipo, è un sistema complesso che assicura il corretto funzionamento di tutto il corpo. L’attuale sequenziamento del genoma rileva alcune mutazioni e varianti che si riscontrano nei tumori. Un team di ricercatori dell’Università di Miami ha scoperto che il sequenziamento dell’intero genoma rileva nuovi cambiamenti legati allo sviluppo del cancro. La ricerca ha evidenziato che sono presenti altri eventi chiave che svolgono un ruolo fondamentale in molti tumori, come: la cromotripsi ed il riarrangiamento genico. Si è scoperto che, con il sequenziamento dell’intero genoma, è possibile leggere tutte le mutazioni nelle regioni proteiche codificanti e non del genoma, nonché le variazioni strutturali.
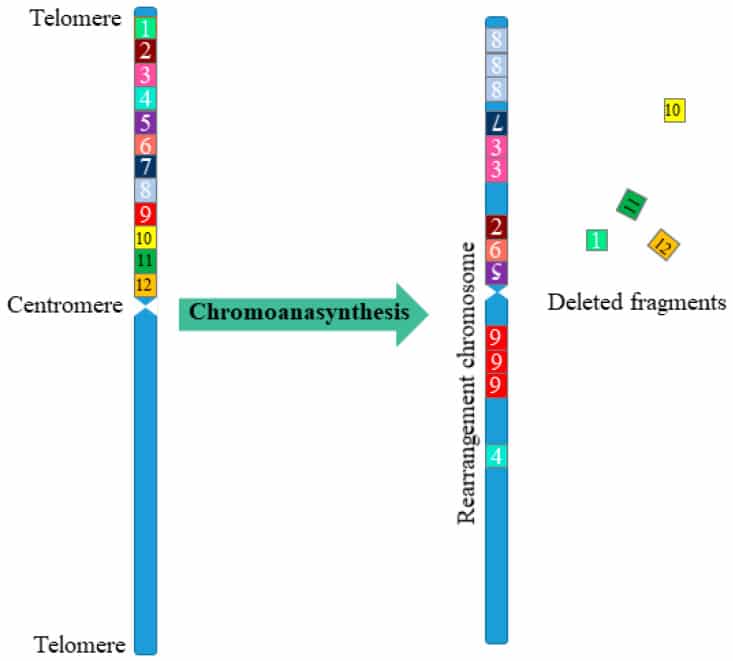
Con il termine riarrangiamento si intende uno spostamento fisico di sequenze del genoma presenti all’interno del nucleo cellulare; quando queste nuove combinazioni interessano i cromosomi si parla di riarrangiamento cromosomico, mentre quando interessano i geni prendono il nome di riarrangiamento genetico. Le modalità del riarrangiamento sono le stesse in ambedue le tipologie. La differenza è nella denominazione delle sequenze di paia di basi.
La cromotripsi è un complesso riarrangiamento cromosomico e si caratterizza da migliaia di riarrangiamenti cromosomici a grappolo che si verificano simultaneamente e si localizzano in regioni limitate del genoma in uno o più cromosomi. Ciò è alla base della formazione di molteplici tipologie di cancro. Questo fenomeno si è scoperto recentemente, nel 2011, ed ha cambiato il concetto di: variabilità del genoma, meccanismi di trasformazione oncogenica e malattie ereditarie.
La ricerca del team ha portato alla creazione di un sistema di smistamento per isolare e ampliare le cellule difficili da studiare e rare di Hodgkin e Reed Sternberg (cHL), i tratti distintivi del cancro del sistema linfatico. La ricerca sui globuli bianchi anormali, se combinata con il sequenziamento dell’intero genoma, ha il potenziale per fornire agli scienziati un modello più dettagliato di alcuni tumori. Il linfoma di Hodgkin è una tipologia di malattia incredibilmente complessa ed, attualmente, c’è ancora molta strada da fare prima di capirla appieno. Sfruttando il sequenziamento dell’intero genoma, è possibile valutare meglio l’evoluzione del tumore, identificandone i problemi strutturali e, ottenendo nuove intuizioni terapeutiche.
Recentemente il National Institutes of Health (NIH) ha comunicato lo sviluppo di un nuovo software innovativo che può assemblare un’intera sequenza del genoma in pochi giorni. Prende il nome di Verkko (“rete” in finlandese) ed è stato creato dal consorzio Telomere-to-Telomere (T2T) con un finanziamento del National Human Genome Research Institute (NHGRI) presente anche nella scoperta del sequenziamento completo del primo genoma umano nel 2022.
Si è preso tutto ciò che si era scoperto precedentemente nel progetto di T2T e si automatizzato il processo. Ora con Verkko, essenzialmente, si può premere un pulsante e ottenere automaticamente una sequenza completa del genoma. Ci sono voluti due decenni per passare dal 92% al 100% nella mappatura del genoma umano, a seguito dell’ambizioso piano del Progetto Genoma Umano per identificare più di tre miliardi di coppie di basi che formano il DNA; inoltre, sono serviti quasi tre anni per assemblare, da parte di T2T, manualmente i difficili frammenti finali per completare il primo genoma umano. Attualmente, Verkko può completare l’attività in pochi giorni generando sequenze geniche senza lacune. Questo nuovo software renderà l’assemblaggio di sequenze geniche complete il più conveniente possibile ed un processo di routine.
È probabile che le regioni appena mappate del genoma umano portino ad una comprensione migliore dei tumori e ad un trattamento personalizzato e più efficace della malattia. Gli scienziati sperano che Verkko possa accelerare le mappature geniche complete di specie di ricerca comuni, come: topi, moscerini della frutta e pesci zebra. Questo nel tentativo di comprendere meglio questi animali complementari. La mappatura delle specie in futuro aiuterà anche nella genomica comparativa e porterà a nuovi legami genetici tra specie apparentemente diverse. Lo sviluppo di Verkko è ancora in corso, con il software in versione beta testing.
{kind=link}
{kind=link}
{kind=link}
{kind=link}