Il progetto PrISMA per il riconoscimento di biomarcatori proteomici

PH: h-its.org
Uno dei metodi più diffusi di identificazione di proteine, chiamato spettrometria MS-MS, include un problema di string matching (confronto di stringhe) computazionalmente intenso. La procedura prevede la ricerca di una lista di piccoli frammenti (peptidi) nell’intero database proteomico umano, al fine di identificare la proteina che essi compongono. La complessità di questo problema cresce sia con la lunghezza della stringa ricercata sia con quelle di riferimento, nonché con il numero di frammenti.
Questa funzione richiede decine di secondi per essere completata e, se eseguita su un GPP (General Purpose Processor), questo lungo tempo di esecuzione si traduce in un’alta richiesta energetica che influisce negativamente sulla scalabilità e sui costi di mantenimento del sistema. Questa problematica può riguardare l’uso dell’applicazione in installazioni su larga scala, come possono essere centri medici o di ricerca che lavorano nell’ambito della proteomica del siero. Inoltre, in questo ambito sono attive varie ricerche orientate al monitoraggio di patologie come, ad esempio, il cancro allo allo stomaco¹, che stanno ottenendo risultati promettenti e che faranno, probabilmente, accrescere il numero di esami richiesti, anche per applicazioni cliniche.

È in questo contesto che si inquadra il progetto PrISMA (Protein Identification through String Matching Algorithm) che Gea Bianchi e Fabiola Casasopra del Politecnico di Milano, in collaborazione con il NECSTLab, stanno portando avanti.
PrISMA punta a velocizzare la fase di string matching del riconoscimento di biomarcatori proteomici e a diminuire i costi energetici, delocalizzando il compito computazionalmente intenso su un cluster di FPGA (Field Programmable Gate Array). I dispositivi utilizzati nel progetto fanno parte del kit AVNET Zedboard.
LA NASCITA DEL PROGETTO
“PrISMA è un progetto nato ad inizio 2015 –racconta a Close-up Engineering Gea Bianchi-, mentre riflettevamo sul nostro lavoro di tesi triennale in Ingegneria Biomedica al Politecnico di Milano. A quel tempo eravamo entrambe studentesse del NECSTLab. Eravamo diventate parte di un laboratorio informatico seguendo il corso del primo anno di informatica tenuto dal prof. Marco D. Santambrogio. In questo contesto abbiamo avuto l’opportunità di ragionare su quale tipo di contributo potessimo apportare nel nostro ambito (l’Ingegneria Biomedica) ed al contempo migliorare le nostre competenze in ambito informatico. Abbiamo ricercato ed analizzato molte possibilità, dall’analisi di immagini (fMRI, Raggi X) all’analisi delle espressioni del viso, pensando anche ad applicazioni in real time (ad esempio, da usare durante gli interventi chirurgici). Durante la nostra ricerca siamo rimaste colpite da un’applicazione che ci permettesse di intersecare in modo bilanciato i nostri interessi: la proteomica del siero e il relativo metodo chiamato shotgun proteomics.”
Il metodo di spettrometria di massa tandem (o spettrometria MS-MS) basato sul metodo shotgun proteomics è una potente tecnologia per l’identificazione di proteine, sia individualmente che in campioni. Recentemente è diventato uno degli approcci più comuni per riconoscere proteine all’interno di una miscela complessa. Questo è esattamente il tipo di approccio di cui si ha bisogno quando si vuole ritrovare una proteina biomarcatore all’interno di un fluido biologico.
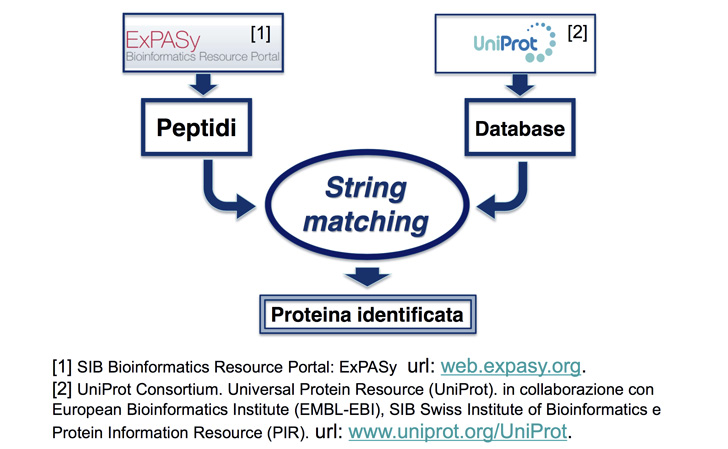
“All’inizio non eravamo ancora esperte nell’ambito –spiegano le ricercatrici-, così decidemmo di continuare ad analizzare la proteomica del siero e la spettrometria di massa tandem. Scoprimmo che questa applicazione era in crescita e trovammo sbalorditivi i suoi domini applicativi, permettendo ai medici di valutare l’andamento di gravi patologie, come possono essere il cancro al seno e allo stomaco, patologie epatiche o l’Alzheimer, nonché le risposte ai trattamenti con un significativo anticipo rispetto alle altre tecniche note (si parla di mesi o anche anni, in base alla singola applicazione). Inoltre, scoprimmo che la fase di string matching (ricerca di sottostringhe –vari frammenti della proteina- all’interno di una stringa, nel database proteomico) è la fase computazionalmente più intensa ed un problema appartenente alla bioinformatica che potrebbe essere migliorato significativamente. Cosa potevamo chiedere di meglio se non sfruttare la nostra tesi come opportunità per migliorare una tecnologia così meravigliosa?
Così è nato il progetto. Da allora, abbiamo lavorato per progettare un design che permettesse un riconoscimento più rapido delle proteine e che ci permettesse di abbattere l’alta richiesta energetica dei costi dell’analisi. Decidemmo quindi di creare una applicazione versatile, che potesse essere usata anche in caso di variazioni del database e della lista di frammenti da ricercare, in modo che rimanesse utilizzabile in ogni applicazione. Inoltre, in questo modo il nostro design potrà essere aggiornato con le nuove proteine revisionate e scoperte ogni anno.
Abbiamo sviluppato la nostra idea con la collaborazione ed il sostegno di dott. Gianluca Durelli e prof. Marco D. Santambrogio.”
DESIGN E RISULTATI
Il primo design è caratterizzato da una configurazione a 4 core implementata su una scheda FPGA Xilinx Zedboard. Il sistema è stato testato e validato con dati reali (una parte di database proteomico umano e il biomarcatore chiamato Ki-67) ed ha rivelato un miglioramento in efficienza energetica con un fattore 6x rispetto ad una implementazione software su un processore Intel i7. Questi risultati sono stati presentati come poster alla conferenza BioCAS 2015².

“Questa è anche stata un’opportunità per visitare per la prima volta gli USA –conclude Gea- e partecipare in prima persona ad una conferenza internazionale, presentando un lavoro di cui siamo estremamente fiere.
Ad oggi stiamo lavorando su PrISMA; l’idea di base dietro questo lavoro è il guadagno di maggiori vantaggi nella computazione grazie all’esecuzione del nostro sistema su un cluster di FPGA. Questa soluzione ci permetterà di salvare l’intero database sulle BRAM delle schede FPGA e di parallelizzare ancor di più il calcolo. Crediamo, in questo modo, di poter rendere l’intero sistema più scalabile ed adatto per installazioni di grande scala”.
Le due ricercatrici parteciperanno allo Xilinx Open Hardware Contest 2016 (#XOHW16), in concomitanza con altri gruppi del NECSTLab del Polimi.



¹ W. Liu, Q. Yang, B. Liu, and Z. Zhu, “Serum proteomics for gastric cancer,” Clinica Chimica Acta, vol. 431, pp. 179–184, 2014.
² Bianchi, Gea, et al. “A hardware approach to protein identification.” Biomedical Circuits and Systems Conference (BioCAS), 2015 IEEE. IEEE, 2015.
[button color=”” size=”large” type=”” target=”_blank” link=”https://www.facebook.com/PrISMA2016/”]Segui il Progetto PrISMA su Facebook[/button]