PrISMA project for proteomic biomarkers recognition

PH: h-its.org
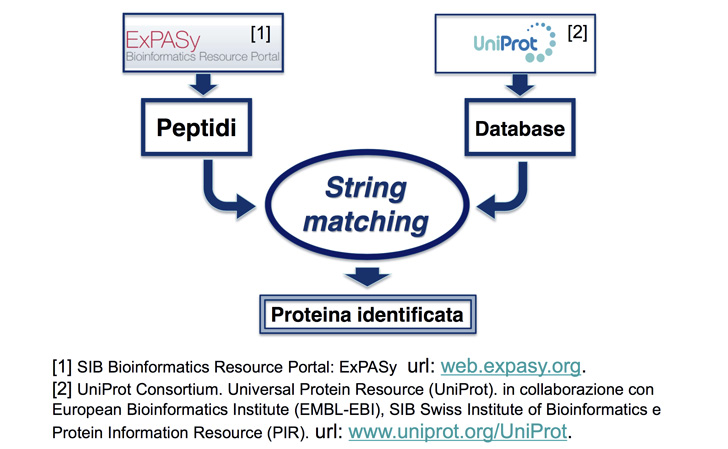
One of the most common protein identification method, called MS-MS Spectrometry, involves a computational intensive string matching problem. The procedure requires searching a list of small fragments (peptides) against the entire human protein database, in order to identify the composed protein. The complexity of the problem grows with the length of both the searched and the reference string and with the number of fragments.
The task takes tens of seconds to complete and, if performed with GPP (General Purpose Processor), this long execution time translates into a high energy requirement, which greatly impacts the scalability and maintenance cost of the system. This problem can affect the use of this application in large scale installations, such as medical or research centers, that work in the serum proteomic field. Moreover, within this field there are many researches oriented to cancer monitoring, such as gastric cancer¹, that are obtaining promising results and that will increase the amount of exams required, also in clinical application.

It is in this context that the PrISMA project (Protein Identification through String Matching Algorithm) is framed; a project that Gea Bianchi and Fabiola Casasopra of the Polytechnic of Milan, in collaboration with the NECSTLab are carrying out.
PrISMA aims to reduce the string matching stage time of the recognition of proteomic biomarkers and to decrease energy costs, relocating the computationally intensive task of a cluster FPGA (Field Programmable Gate Array). The devices used in the project are part of AVNET ZedBoard kit.
THE BIRTH OF THE PROJECT
“PrISMA is a project started at the beginning of 2015 – says Gea Bianca at Close-up Engineering -, while we were working at our bachelor thesis in Biomedical Engineering at Politecnico di Milano.
Back at that time, we were both students at the NECST Lab. We joined a CS lab because of the course project in a CS101-like class done by Prof. Marco D. Santambrogio. This context gave us the chance of reasoning about which kind of contribution we could bring to our field of studies (biomedical engineering) while strengthening our CS interest and knowledge. We explored many possibilities, from image processing, e.g. image analysis (fMRI, X-Ray) and facial expression identification, to real-time applications, e.g. for surgery needs. While doing this, we got dazzled by an application that was allowing us to perfectly balanced our interests: serum proteomics and the related shotgun proteomics method.”
Shotgun-proteomics-based tandem mass spectrometry (or MS-MS Spectrometry) is a powerful technology for identifying proteins, whether individually or in samples. It has recently become one of the most used approaches to recognise proteins in a complex mixture. That is exactly the kind of approach you need when you are looking for protein biomarkers within a biological fluid.
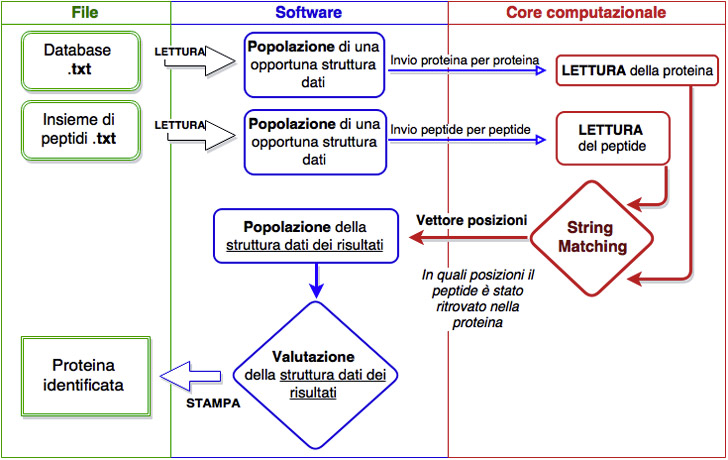
Flux of PrISMA data
“At the very beginning we were not expert of that area and we decided to go further analysing serum proteomics and tandem mass spectrometry. We found out that the applicative domains were this technology could be applied to were just huge and extremely interesting, e.g., allowing doctors to track the development of serious diseases such are gastric cancer, breast cancer, liver diseases or Alzheimer, and patients’ responses to treatments with a significant advance over the other known techniques (they talk about months or even years). Moreover, the string matching phase within the technique was the most computational intensive task and a bioinformatics problem that could be significantly improved. What could be better than using our thesis as an opportunity of improving such a wonderful technology?
That is how the project was born. Since there, we have been working to design a system capable of faster recognition of proteins and that could decrease the high energy costs of the analysis. We decided to create a versatile application that could be used varying both the database and the list of protein fragments to search against it, so that it could remain suitable independently of the specific application. Moreover, in such a way our design could be updated with revised or brand new proteins.
We developed our idea with the collaboration of doc. Gianluca Durelli and prof. Marco D. Santambrogio
DESIGN AND RESULTS
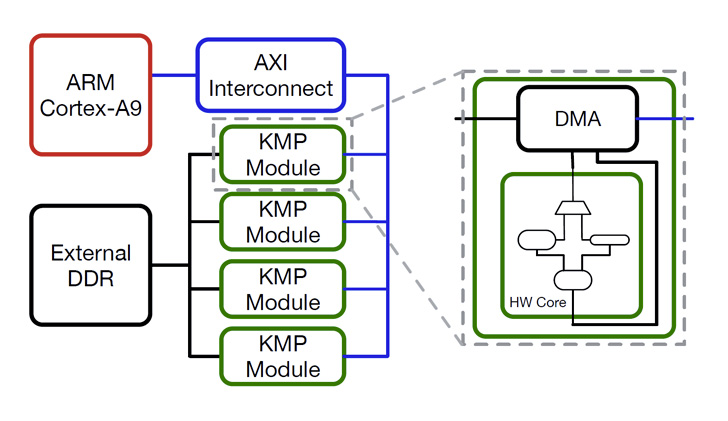
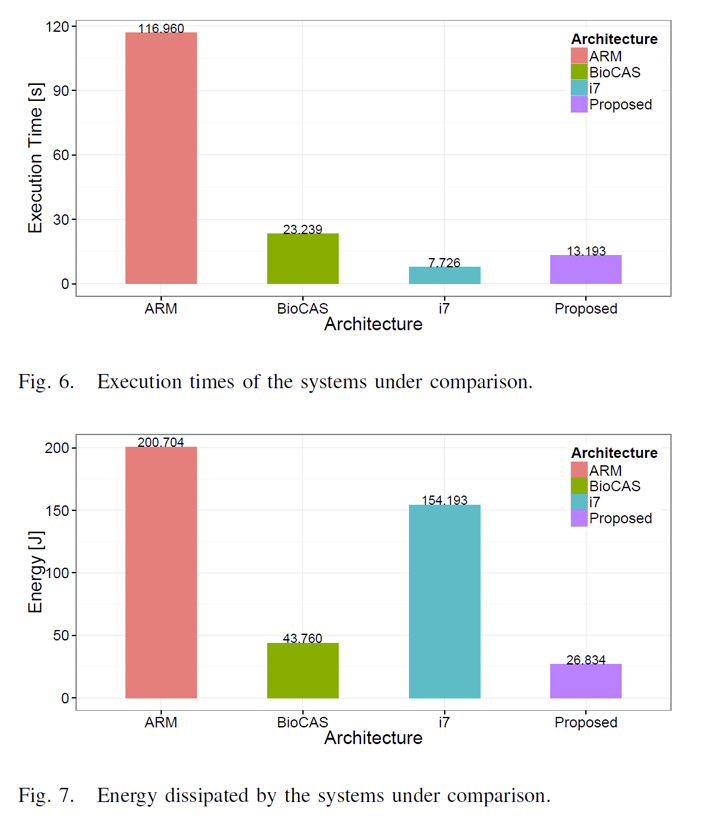
Our first design was characterized by a 4-core system, implemented on a Xilinx Zynq FPGA. It was validated with real world data (part of the human protein database and a biomarker called Ki-67) and it showed a 6X factor of increased energy efficiency with respect to an Intel i7 software implementation. These results have been presented as a poster at the BioCAS 2015 conference2.

“This was also the opportunity for us to visit for the first time the USA – concludes Gea -and to attend an international conference to present a work that we were so proud of.
Right now we are working on PrISMA, the basic idea behind this work is to gain more advantages in the computation by executing our system on an FPGAs cluster. This solution will allow us to store the entire database on the FPGAs BRAMs and to parallelize even more the computation. We believe that in such a way we could make the entire system more scalable and suitable for large-scale installations”.
The two researchers will participate in the Xilinx Open Hardware Contest 2016 (#OHW16), along with other groups of the NECSTLab of Polimi.
¹ W. Liu, Q. Yang, B. Liu, and Z. Zhu, “Serum proteomics for gastric cancer,” Clinica Chimica Acta, vol. 431, pp. 179–184, 2014.
² Bianchi, Gea, et al. “A hardware approach to protein identification.” Biomedical Circuits and Systems Conference (BioCAS), 2015 IEEE. IEEE, 2015.
[button color=”” size=”large” type=”” target=”_blank” link=”https://www.facebook.com/PrISMA2016/”]Follow PrISMA on Facebook[/button]